Philosophical Premise
The Worsening Biomedical Crisis
Biomedical science is in crisis. Investments in health care research and practice are increasing exponentially, while the release of effective new therapeutics declines annually. It is no wonder that the American people are becoming disillusioned with medicine and biomedical research, but what is the problem?
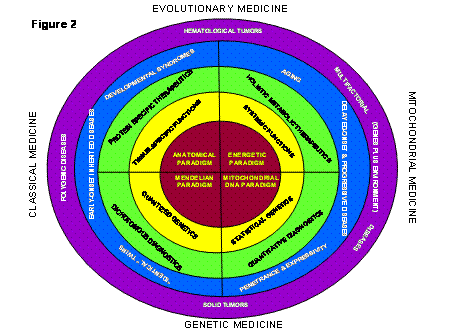
Figure 2: New Biomedical Paradigm that Combines the Traditional Structural Biology and Mendelian Genetics of the Nuclear-Cytosolic Organism with the Energy Biology and Mitochondrial DNA Genetics of the Mitochondrial Organism. The four sectors indicate the different aspects of human biology and genetics determined by the functional and genetic components of the two different organisms.
The Center for Molecular Medicine (CMM) at Emory University and its progeny, the Center for Molecular and Mitochondrial Medicine and Genetics (MAMMAG) at the University of California, Irvine (UCI), were founded on the premise that the fault lies in the prevailing paradigms that guide today’s biomedical research and medical practice (Figure 2).
The prevailing paradigm of medicine is anatomical. If you have a headache, it is assumed that the problem is within your head. This anatomical paradigm traces back 450 years to the Belgian-Italian anatomist, Vesalius, who was the first to accurately describe the physical structures of the human body. His efforts and those that followed have resulted in anatomy being the first course taken by all medical students, the establishment of organ-specific departments and divisions within medical schools and hospitals (e.g. neurology, ophthalmology, nephrology, cardiology, endocrinology, etc), and the preoccupation of drug companies with developing organ-specific therapeutics (Figure 2, upper left hand quadrant).
The anatomical paradigm directs biomedical researchers to study the anatomy of the brain to understand Alzheimer Disease (AD) and Parkinson Disease (PD) and the structure of the heart and coronary arteries to understand cardiovascular diseases. It directs molecular biologists to clone tissue- and organ-specific genes to determine the molecular basis of organ-associated symptoms and guides the molecular pharmacologist to look for chemicals that bind to tissue-specific proteins in hopes of treating organ-associated symptoms (Figure 2, upper left hand quadrant).
However, most of the genes in the human genome are not tissue-specific. Rather, they are systematic, being expressed in a wide variety of tissues but at different levels. Since various organs rely on the range of systemic genes to different extents, defects in systemic genes can cause organ-associated symptoms.
In classical biomedical science, the prevailing anatomical paradigm of medicine is paired with the prevailing Mendelian paradigm of genetics (Figure 2, lower left hand quadrant). Originally defined by Gregor Mendel almost 150 years ago and championed by Drosophila geneticists in the early twentieth century, the Mendelian genetic paradigm argues that all genes occur in the body in two copies, one contributed by the father and the other by the mother when the sperm and egg (oocyte) fuse at conception. At sexual maturity, the two gene copies are again segregated into the individual sex cells during meiosis. These rules of Mendelian genetics are mirrored in the behavior of the chromosomes of the nucleus and thus apply to the inheritance of the nuclear DNA (nDNA) (Figure 2, lower left hand quadrant).
The Mendelian genetic paradigm makes powerful predictions. All genetic traits must be quantized into three states (two healthy genes: +/+; one healthy and one damaged: +/-; or two damaged: -/-), genetic traits must be inherited in families in specific patterns, and all of the genes must be identical in identical twins while only half of the genes must be identical in fraternal twins. As a corollary of these principles, it is assumed that if a human clinical trait is more concordant among identical twins than fraternal twins, it has a genetic component, but if the trait is equally concordant in identical and fraternal twins, then it is due to environmental influences (Figure 2, lower left hand quadrant).
However, many clinical phenotypes that are clearly familial do not follow the rigid rules of Mendelian inheritance. Examples include late-onset AD and PD, migraines, cardiovascular disease, diabetes, obesity, cataracts and macular degeneration, and cancer. Starting from the Mendelian paradigm, such diseases are assumed to result from the interaction of more than one Mendelian gene, and are thus called “polygenic traits” (Figure 2, left). Based on this extrapolation, if the clinical molecular geneticist cannot find one gene that accounts for the phenotype, then it is assumed that the phenotype is caused by two gene defects, if not two then three, ad infinitum. Because of this artifice, any familial trait can be argued as having a Mendelian basis. In fact, in most cases, the common age-related diseases cannot be explained by one of a few "Mendelian" genes. Chromosomal genetic variants explain less than 10 to 20% of cardiovascular disease, AD, PD, breast cancer, prostate cancer, colon cancer, and have provided no meaningful explanation for aging. This difficulty has led to the assumption that these diseases are caused by a large number of Mendelian genes each contributing a small portion of the disease risk. To find such putative genes has required the screening of increasingly large populations of patients and controls with ever growing numbers of chromosomal gene genetic markers, yet with continually diminishing returns.
Paradigm Crises in Science
This type of scientific crisis is not new. Based on the prevailing paradigm that the Earth was the center of the universe and that the Sun revolved around the Earth, Ptolemy evoked a complex and elegant mathematical model involving a celestial sphere along which the various heavenly bodies moved. The periodic retrograde motion that was observed for the planets was rationalized by having the planets move around epicycles whose vertices followed the celestial spheres. This model of the heavens was accepted for a thousand years until Galileo, Kepler and Copernicus showed that the motion of the planets could be explained much more simply by having the Earth and the other plants revolve around the Sun. Ptolemy’s model could not be generalized beyond the solar system, but the Kepler-Copernicus model has been generalized to the entire known universe. Hence, a change in a limited prevailing paradigm can open the way for a new wave of scientific discoveries and advances.
In his classic treatise on “The Structure of Scientific Revolutions (1962)“ (3rd Edition, 1996, U. Chicago Press), Thomas S. Kuhn envisioned that science advanced in a series of steps: the collection of facts, the summarization of the facts into theories, the testing of theories by hypothesis-driven experimentation (“normal science”), and the elevation of the theory to a prevailing paradigm (“law”) based on repeated demonstration of its predictive power. The problem with paradigms is that they are human constructs based on the limited set of facts to be explained. Therefore, human paradigms are frequently incomplete and eventually encounter phenomena outside the purview of the paradigm. At this point investment in predictions of the paradigm becomes increasingly unproductive and scientific progress stalls. According to Kuhn the only way to resolve such a scientific paradigm crisis is to recast the old paradigm into a new paradigm which not only explains the previous phenomena, but has new elements that can make verifiable predictions about the unexplained phenomena.
We believe that we are now at such a paradigm crisis in the biomedical science in which the clinical phenomena to be addressed now lie predominantly outside the purview of the prevailing structural and Mendelian paradigms (Figure 2, left half). As a result, purveyors of the prevailing paradigms have been forced to devise an increasing number of ad hoc assumptions in a vain attempt to rationalize the observed characteristics of age-related diseases, aging, and cancer.
The CMM and MAMMAG were founded on the premise that Center scientists have discovered a crucial subset of the missing elements of the anatomical and Mendelian paradigms. These elements reside in mitochondrial energy metabolism and mitochondrial genetics (Figure 2, right side).
Two Organisms Make a Man
Humans are limited by their individual 'I' perspective, which is singular. Yet we know that each person is made of 30 trillion individuals called cells. Hence the 'I' is really a 'we'. This idea is also myopic, however, for we now know that each nucleated (eukaryotic) cell is composed of two different life forms that formed a symbiosis three to four billion years ago. Today, these two cells are represented by the nuclear-cytosol “host” cell and a colony of cytosolic bacteria called “mitochondria.” Thus, inside each of the 'we's is a 'they' (Figure 3).

Figure 3: The Cellular Structures of the Two Human Organism and Their Symbiotic Relationship. The left panel shows the relative physical relationship of the nucleus-cytosol and the mitochondria. The right upper panel shows a high resolution picture of the mitochondrial organism and the right lower panel shows electron micrographs of the mitochondrial DNA (mtDNA).
These two organisms were co-equal at the time of the original symbiosis. However, over the intervening billions of years they have become specialized. The nuclear-cytosol organism specialized in generating the structures of the cells and later of the tissues and organs (Figure 2, left half). The mitochondria specialized in energy generation to fuel the development of these increasingly complex structures, yet they still retain their own genomic integrity and separate information storage and retrieval systems (Figure 2, right half).
Until now, biomedical science has only considered the nuclear-cytosol organism in relation to disease. It is this organism’s genes that follow the Mendelian rules of inheritance and choreograph development determining the complex structures of the human body. However, to power development and fuel the building and animation of the structures requires energy and this is the purview of the mitochondrion (Figure 2, upper right quadrant). Moreover, the genes of the mtDNA are distinct, being present in thousands of copies per cell and are inherited exclusively from the mother (Figure 2, lower right quadrant).
The relevance of the mitochondria, the other human cell which controls energy and has non-Mendelian genes, to clinical medicine remains virtually unexplored by biomedical science. Hence, mitochondrial energetics and mitochondrial genetics could provide the crucial missing pieces of the structural and Mendelian paradigms required to explain the puzzling features of age-related diseases.
The Mitochondrial Paradigm for Common Diseases
Life is structure plus energy. The cadaver dissected by the first year medical student is dead, not the patient’s desired outcome when entering the doctor’s office. What differentiates the cadaver from the patient, animation and thus energy [1](Figure 4)?
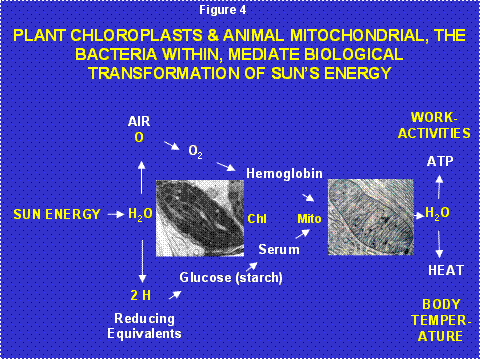
Figure 4: Transformations of the Sun’s Energy by the Plant Cell Symbiotic Chloroplast and the Animal Cell Symbiotic Mitochondria. The chloroplast, derived from cyanobacteria, uses the Sun’s energy to split water releasing the oxygen in the atmosphere and fixing the hydrogen to carbon to generate glucose. Animals eat the plants to acquire the glucose and collect the oxygen from the atmosphere by breathing. Their mitochondria remove the hydrogen from the hydrocarbons and react it with the oxygen to regenerate water, releasing the Sun’s energy to generate ATP to drive work and heat to maintain body temperature.
Virtually all terrestrial biological energy is derived from the Sun. Plants collect the Sun’s photons and use the energy to split water into hydrogen and oxygen. The hydrogen is attached to carbon to create the simple sugar glucose. Excess glucose is stored as starch. The oxygen is released into the atmosphere as O2. The prokaryotic (before the nucleus) cyanobacteria invented photosynthesis and perform it within the internal membranes of their cells. The original “eukaryotic” cell with its combination of nuclear-cytosol and mitochondrial organisms could not perform photosynthesis (Figure 3). However, some of the early eukaryotic cells also incorporated cyanobacteria as symbionts giving rise to the chloroplasts of multicellular plants. Hence, all of the glucose and starch of higher plants is generated by bacteria-derived chloroplasts (Figure 4).
Animals including humans eat plants to acquire the Sun’s energy in the form of starch and thus glucose. The hydrogen attached to these hydrocarbons is removed by the mitochondrion and reacted with the oxygen breathed from the atmosphere to regenerate H2O, releasing the Sun’s energy to power our bodies. Hence, our mitochondria are the power plants of our cells and we eat and breathe to stoke our mitochondrial furnaces (Figure 4).
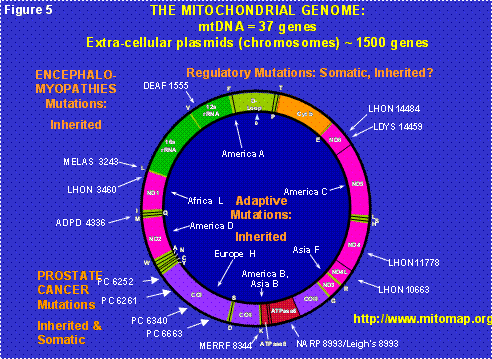
Figure 5: The Human Mitochondrial DNA Map. D-loop = control region. Letters around the outside perimeter indicate cognate amino acids of the tRNA genes. Arrows followed by continental names and associated letters on the inside of the circle indicate the position of defining polymorphisms of selected region-specific mtDNA lineages. Arrows associated with abbreviations followed by numbers around the outside of the circle indicate representative pathogenic mutations, the number being the nucleotide position of the mutation. The abbreviations are DEAF = deafness; MELAS = mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes; LHON = Leber’s hereditary optic neuropathy; ADPD = Alzheimer and Parkinson disease; MERRF = myoclonic epilepsy and ragged red fiber disease; NARP = neurogenic muscle weakness, ataxia, retinitis pigmentosum; LDYS = LHON + dystonia.
Each cell contains hundreds of mitochondria and each mitochondrion contains multiple copies of its own DNA, the mtDNA. Hence, each cell contains thousands of mtDNAs (Figure 3). The original bacterial genome contained all of the genes necessary to make a free living bacterium. However, the bacteria within the eukaryotic cell’s cytosol must replicate fast enough to assure that when the cell divides (cytokinesis) some of the progeny bacteria will colonize each of the daughter cells. Therefore, the mitochondria are under strong selective pressure to replicate fast, and thus to have as small a DNA as possible (Figure 5). Because of this, the mitochondria initially lost all of their genes for independent living. Then they invented a system by which they could transfer about 1500 of their genes to extracellular plasmids, the nDNA of the chromosomes, where these mitochondrial genes are now replicated, transcribed into cytosolic mRNAs which are translated on cytosolic ribosomes. The resulting proteins are transported back into the mitochondrion.
Today, the human mtDNA is a circular molecule of 16,569 nucleotide pairs (nt) that encodes the small and large rRNA subunits for the mitochondrial ribosome, the 22 tRNAs for mitochondrial translation, and 13 polypeptides. These 13 mtDNA polypeptides are very special for they are all essential components of the multi-polypeptide enzyme complexes of oxidative phosphorylation (OXPHOS), the mitochondrial energy generating system (Figure 5).
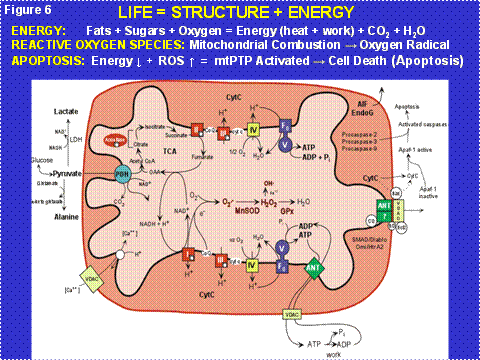
Figure 6: Mitochondrial Energy Production and Its Reaction to the Pathophysiology of Disease. Three features of mitochondrial metabolism are central to the physiology of the common age-related diseases: (1) energy production by oxidative phosphorylation (OXPHOS); (2) reactive oxygen species (ROS) generation as a toxic by-product of OXPHOS; and (3) regulation of apoptosis through activation of the mitochondrial permeability transition pore (mtPTP). ADP or ATP = adenosine di- or tri-phosphate; ANT = adenine nucleotide translocator, cytc = cytochrome c; GPx = glutathione peroxidase-1; LDH = lactate dehydrogenase; MnSOD = manganese superoxide dismutase or SOD2; NADH = reduce nicotinamide adenine dinucleotide; TCA = tricarboxylic acid cycle; VDAC = voltage dependent anion channel. I, II, III, IV, and V = OXPHOS complexes I-V. Complex I is composed of 46 polypeptides, seven (ND1, 2, 3, 4L, 4, 5, 6) encoded by the mtDNA; complex II consists of four nDNA encoded polypeptides; complex III by 11 polypeptides, one (cytb) encoded by the mtDNA; complex IV is composed of 13 polypeptides, three (COI, II, III) encoded by the mtDNA; and complex V is composed of 16 polypeptides, two (ATP6,8) encoded by the mtDNA.
The mitochondria recover the hydrogen from the glucose and then conduct the electrons from the hydrogen down a biochemical wire (redox gradient), the Electron Transport Chain (ETC). The ETC encompasses the multiple polypeptide complexes I, II, III, and IV (Figure 6). The ETC ends with cytochrome c oxidase (COX, complex IV) which combines the electrons with oxygen to give water. The energy that is released as the hydrogen is progressively oxidized is used by the OXPHOS enzyme complexes I, III, and IV to pump positive charges (protons) out across the mitochondrial membrane to generate a capacitor that is negative on the inside and positive on the outside of the mitochondrial inner membrane. The potential energy stored in this capacitance can then be used for many purposes including the synthesis of ATP (the chemical energy currency used to do work) by complex V; heat to maintain our body temperature at 37°C; the transport of substrates and ions including Ca++; etc.
As a toxic by-product of mitochondrial combustion, the mitochondria generate oxygen radicals including superoxide anion (O2-), hydrogen peroxide (H2O2), and hydroxyl radical (OH), collectively known as reactive oxygen species (ROS); the smoke of the mitochondrial furnace (Figure 6). This ROS damages mitochondrial lipids, proteins and DNA. As a partial consequence of ROS damage, the mtDNA has a very high mutation rate, on the order of twenty times that of nuclear genes with comparable function.
Since the mtDNA is in the cytoplasm and thus outside the chromosomes of the nucleus, it is not inherited according to the laws of Mendel. In fact, the mtDNA is inherited only through the oocyte and thus is exclusively maternally inherited (Figure 7). Since each cell contains thousands of copies of each mtDNA gene, a mutation in a mtDNA creates an intracellular mixture of mutant and normal mtDNAs, a state called heteroplasmy. As the cell divides, the percentage of mutant and normal mtDNAs can drift by chance along the radiating cell lineages, a process called replicative segregation (Figure 7). As a result, over many cellular mitotic or meiotic divisions, the percentage of mutant mtDNAs can drift from 1% mutant, to 10, to 25, to 50, to 75, to 100%. As the percentage of mutant mtDNAs increases the mitochondrial energy output declines, the cellular equivalent of a metropolitan brown-out. When the cell lacks sufficient energy to perform its function, its bioenergetic threshold, the mitochondrion senses the energy deficiency and activates an internal self destruction system, the mitochondrial permeability pore (mtPTP) composed of the ANT, VDAC, Bax, Bal-2, etc. (Figure 6, right side spanning the inner and outer membranes). This destroys the energy deficient malfunctioning cell so that it does not interfere with the function of adjacent normal cells. The problem for modern medicine is that our post-mitotic tissues have a finite number of cells. Since mitochondrial energy production must go on throughout life, cells die over time. If we live long enough, sufficient cells are lost from one or more tissues to result in organ malfunction and clinical symptoms.
We first discovered inherited pathogenic mtDNA mutations in 1988, when we reported the mtDNA mutation that cause Leber’s Hereditary Optic Neuropathy (LHON), a form of mid-life blindness [2], and subsequently the mutation that causes Myoclonic Epilepsy and Ragged Red Fiber Disease (MERRF), a progressive epilepsy with muscle weakness [3][4]. Since then, a large number of mtDNA mutations have been linked to familial symptoms, including forms of blindness, deafness, dementia, stroke-like episodes, migraines, movement disorders, cardiomyopathy, renal dysfunction, diabetes, various forms of cancer, etc. Since mtDNA mutations are distributed to every cell of the body, systemic mtDNA defects can and do cause organ-associated symptoms (Figure 2, upper right quadrant). Moreover, different percentages of the same mtDNA mutation can cause different clinical symptoms. The mtDNA tRNALeu(UUR) nt A3243G mutation results in a systemic defect in mitochondrial protein synthesis. However, patients that harbor 10 to 30% mutant mtDNAs develop Type II diabetes and/or migraines and deafness, while patients that harbor over 75% mutant mtDNAs can develop cardiomyopathy, or stroke-like episodes, or ophthalmoplegia. Moreover, identical twins that receive different percentages of a mutant mtDNA develop totally different clinical phenotypes and identical and fraternal twins have an equal probability of inheriting the same mutant mtDNA genotype and thus exhibiting the same clinical phenotype. Thus, phenotypic differences are equally likely to be concordant in identical versus fraternal twins and may not be due to environmental causes but rather due to heteroplasmic mtDNA mutations (Figure 2, lower right quadrant)[1].
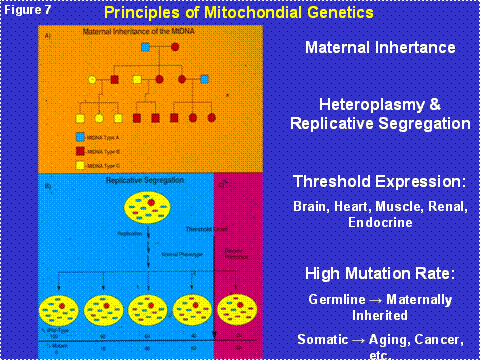
Figure 7: The Principles of Mitochondrial DNA Genetics. Upper Panel = maternal inheritance. Lower Panel = mtDNA heteroplasmy, replicative segregation, and bioenergetic threshold leading to cellular energetic failure and death.
mtDNA mutations also accumulate with age in post-mitotic tissues such as the brain, heart, muscle and kidney. This is because the mtDNA continues to replicate in post-mitotic cells even though the nDNA becomes dormant in G0 of the cell cycle. The accumulation of these “somatic mtDNA mutations” erodes mitochondrial function throughout life until sufficient cells die that organs malfunction resulting in the symptoms of old age (Figure 8). The primary source of these mtDNA mutations is ROS damage[1]. This was proven when we showed that increasing the mitochondrial antioxidant defenses, either chemically or by genetic manipulation, blocked the onset and progression of cardiomyopathy and neurological disease and extended the life span in both nematodes and mice [5]. Thus, the accumulation of somatic mtDNA mutations in post-mitotic tissues is the aging clock [1] (Figure 2, right half).
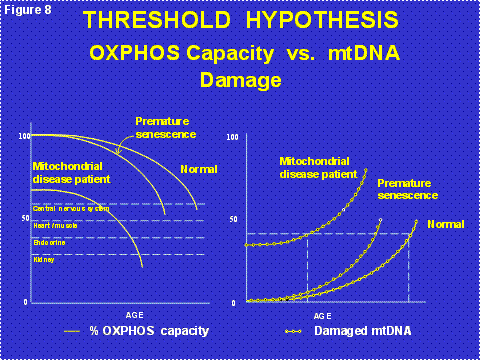
Figure 8: The Threshold Hypothesis of the Clinical Expression of Mitochondrial Energy Deficiency. Left Panel = the age related decline in mitochondrial energy generating capacity showing the starting energetic levels of individuals with initially highly functional mitochondria (upper lines) or partially dysfunctional mitochondria (lower line). The upper line is shown to decay with two different kinetics: slowly resulting in normal aging or more rapidly, possibly due to increased mitochondrial reactive oxygen species (ROS) production, resulting in “premature” development of symptoms as might be seen for a patient with Type II diabetes, Alzheimer Disease, Parkinson Disease, cardiovascular disease, or ophthalmological deterioration. Right Panel, shows the relative proportion of mutant mtDNAs with age showing the cumulative effects of inherited plus somatic mtDNA mutations which lead to the erosion of mitochondrial energetic function.
Since the milder mtDNA diseases have a delayed onset and a progressive course, we now believe that the clinical symptoms of the common “polygenic” diseases arise from the interaction between the inheritance of nDNA and/or mtDNA mutations which cause partial mitochondrial defects which are subsequently exacerbated by the progressive accumulation of somatic mtDNA mutations [6] (Figure 8). When the combination of inherited and somatic mutational defects become sufficient to reduce mitochondrial energy production below the bioenergetic threshold, cells are lost. Since we are only born with sufficient cells to optimize our reproductive success, and fertility ends at age 50 in the human female, our tissues begin to run out of enough cells to maintain normal function as we age beyond 50 years [1](Figure 9).
While excessive mitochondrial ROS production is toxic to the cell, low levels of mitochondrial ROS are mitogenic. One hypothesis is that the nucleus uses mitochondrially-generated H2O2 to monitor mitochondrial number. Since more mitochondria would make more ROS, when the nucleus perceives excessive ROS it is programmed to enter mitosis. Cell division cuts the number of mitochondria in half, thus restoring the proper nuclear-mitochondrial ratio.
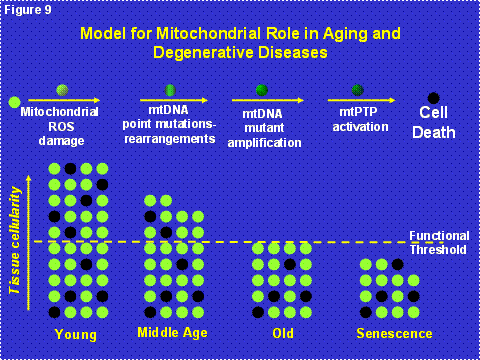
Figure 9: Mitochondrial and Cellular Model of Aging. The upper line of cells diagrams the mitochondrial role in the energetic life and death of a cell. All post-mitotic cells have a finite probability of dying due to the accumulation of mitochondrial and mtDNA damage associated with OXPHOS ROS production, followed by mutant mtDNA amplification, activation of the mtPTP, and death by apoptosis. The bottom diagram represents the loss of cells in a tissue over the life of an individual through mitochondrial mediated death, black cells. Each individual is born with sufficient extra cells such that even as cells are lost by mitochondrial induced apoptosis, enough cells remain to maintain tissue function. Ultimately, however, sufficient cells are lost that the organ begins to malfunction resulting in the clinical manifestations of old age. The minimum number of cells for the tissue to function normally is indicated by the yellow dashed line.
However, a partial defect in mitochondrial OXPHOS caused by a mild mtDNA mutation can impede the energy production process. This also results in increased ROS production, comparable to throwing water onto a fire causing partial combustion, reducing heat and light, and increasing smoke. Therefore, certain mtDNA mutations that increase ROS production act as mitogens, driving the cell into mitosis, a key step in cancer. Consistent with this hypothesis, mtDNA mutations are increasingly being recognized as a central feature of cancer biology [7][8] (Figure 2).
Since the mitochondria convert the calories available from our environment into energy which must be apportioned into either heat to maintain our body temperature or ATP to perform work, and the mtDNA encode the key genes that determine how the energy is apportioned, mtDNA mutations have been a major adaptive mechanism permitting humans to adapt to new environments by modifying the efficiency of OXPHOS. Since the mtDNA is exclusively maternally-inherited it can only change by the sequential accumulation of mtDNA mutations along radiating maternal lineages. Hence, all mtDNA sequence variants can be fit into a single dichotomous mutational tree which dates back to a founding mtDNA that lived in Africa about 200,000 years before present (YBP) (Figure 10). People were confined to Africa for about 140,000 years, during which the mtDNAs radiated to give four Africa-specific lineages, L0-L3. About 65,000 YBP, however, two new mtDNAs lineages, M and N, arose in northeastern Africa. Only these succeeded in leaving tropical and subtropical Africa to colonize all of temperate Eurasia. A plethora of mtDNA lineages were then spawned from M and N in Eurasia. However, of all of the temperate zone mtDNAs, only four lineages (A, C, D, and X) succeeded in crossing the arctic to colonize the Americas [1] (Figure 10).
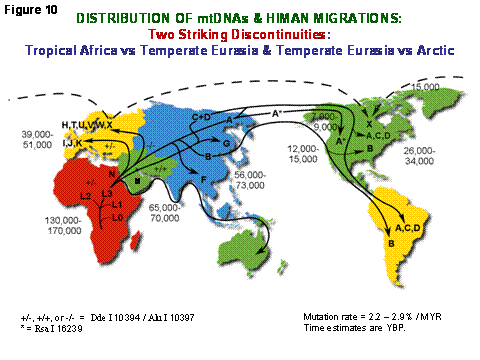
Figure 10. Diagram Outlining the Migratory History of the Human mtDNA Haplogroups. Homo sapiens' mtDNAs arose in Africa about 150,000 to 200,000 years before present (YBP), with the first African-specific haplogroup branch being L0, followed by the appearance in Africa of lineages L1, L2, and L3. In northeastern Africa, L3 gave rise to two new lineages, M and N. Only M and N mtDNAs succeeded in leaving Africa and colonizing all of Eurasia about 65,000 YBP. In Europe, N gave rise to the H, I, J, Uk, T, U, V, W, and X haplogroups. In Asia, M and N gave rise to a diverse range of mtDNA lineages including A, B, and F from N and C, D, and G from M. A, C, and D became enriched in northeastern Siberia and crossed the Bering Land Bridge about 20,000 to 30,000 YBP to found the Paleo-Indians. At 15,000 YBP, haplogroup X came to central Canada either from across the frozen Atlantic or by an Asian route of which there are no clear remnants today. At 12,000 to 15,000 YBP, haplogroup B entered the Americas, bypassing Siberia and the arctic, likely by moving along the Beringian Coast. Next at 7000 to 9000 YBP, a migration bringing a modified haplogroup A moved from northeastern Siberia into northwestern North America to found the Na-Dene (Athebaskins, Dogrib, Apaches, and Navajos). Finally, relatively recently, derivatives of A and D moved along the Arctic Circle to found the Eskimos. These observations revealed two major latitudinal discontinuities in mtDNA variation: one between the African L haplogroups and the Eurasian N and M derivatives and the other between the plethora of Central Asian mtDNA lineages and the almost exclusive presence of lineages A, C, and D in northeastern Siberia, the latter spawning the initial wave of Native American migrations, followed by X and then B. Since these discontinuities correspond to the transitions from tropical and subtropical to temperate and from temperate to arctic, we have proposed that these discontinuities were the result of climatic selection of specific mtDNA mutations that permitted certain female lineages to prosper in the increasingly cold northern latitudes [9].
It is now clear that two major discontinuities exist in mtDNA variation: between tropical and sub-tropical Africa and temperate Eurasia, and between temperate Eurasia and the arctic. Moreover, these discontinuities are the product of the appearance of new functional mtDNA mutations that shifted the OXPHOS energy output from predominately ATP production and very little heat production in tropical Africa to subsequently increased heat production at the expense of ATP production in the temperate and arctic zones. About 25% of all ancient mtDNA sequence variation appears to have been adaptive [10][11]. This same variation is now being discovered by multiple investigators to influence longevity, predisposition to age-related degenerative diseases such as AD and PD, and to influence the clinical expression of the milder nDNA and mtDNA pathogenic gene mutations [1].
Amazingly, many of these same ancient human adaptive mtDNA mutations have also been found to arise as new somatic mutations as cells undergo neoplastic transformation to become malignant cancer cells. This makes a kind of sense since the cancer cell must adapt to new environmental conditions as have humans. In both cases the environmental conditions affect the availability of energy substrates, oxygen tension, and ROS toxicity, all the purview of the mitochondria [8].
A Two Pronged Approach to 21st Century Medicine
Therefore, the discovery of mitochondrial diseases and the elucidation of the rules of mitochondrial genetics have expanded the classical medical paradigms of structural diseases and Mendelian inheritance to energy diseases and mtDNA inheritance (Figure 2). With the addition of the biology of the mitochondrial organism, we now have a conceptual framework that is capable of explaining the natural history and complex inheritance of the complex diseases.
This mitochondrial perspective on complex diseases defines two major classes of therapeutics: interventional and preventative. Interventional therapeutics treat existing symptoms, which result from excess cell loss and thus must treated by replacing the lost cells. The goal of preventional therapeutics is to prevent the onset and progression of symptoms and must focus on preserving existing cells by stabilizing their mitochondrial function. This can be accomplished in three ways: improving mitochondrial energy generation, reducing ROS production, and stabilizing the mtPTP and thus inhibiting apoptosis.
Therefore, since translational research leading to effective treatments has always been the object of CMM and MAMMAG, our research and development efforts fall into two major programs: (1) Mitochondrial and Molecular Medicine and (2) Germ Cell and Stem Cell Biology, known collectively as the Mitochondrial and Germ and Stem Cell Biology Unit (MAGSCEB) (Figure 1, left top quadrant).
Bibliography
- Wallace, D.C., A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annual Review of Genetics, 2005. 39: p. 359-407.
- Wallace, D.C., et al., Mitochondrial DNA mutation associated with Leber's hereditary optic neuropathy. Science, 1988. 242(4884): p. 1427-1430.
- Wallace, D.C., et al., Familial mitochondrial encephalomyopathy (MERRF): Genetic, pathophysiological, and biochemical characterization of a mitochondrial DNA disease. Cell, 1988. 55(4): p. 601-610.
- Shoffner, J.M., et al., Myoclonic epilepsy and ragged-red fiber disease (MERRF) is associated with a mitochondrial DNA tRNALys mutation. Cell, 1990. 61(6): p. 931-937.
- Schriner, S.E., et al., Extension of murine life span by overexpression of catalase targeted to mitochondria. Science, 2005. 308(5730): p. 1909-1911; published online 5 May 2005 (10.1126/science.1106653).
- Coskun, P.E., M.F. Beal, and D.C. Wallace, Alzheimer's brains harbor somatic mtDNA control-region mutations that suppress mitochondrial transcription and replication. Proceedings of the National Academy of Sciences of the United States of America, 2004. 101(29): p. 10726-10731.
- Petros, J.A., et al., mtDNA mutations increase tumorigenicity in prostate cancer. Proceedings of the National Academy of Sciences of the United States of America, 2005. 102(3): p. 719-724.
- Wallace, D.C., Mitochondria and Cancer: Warburg Address. Cold Spring Harbor Symposium, 2006. 70: p. In press.
- Wallace, D.C., The mitochondrial genome in human adaptive radiation and disease: On the road to therapeutics and performance enhancement. Gene, 2005. 354: p. 169-80. 10. Mishmar, D., et al., Natural selection shaped regional mtDNA variation in humans. Proceedings of the National Academy of Sciences of the United States of America, 2003. 100(1): p. 171-176. 11. Ruiz-Pesini, E., et al., Effects of purifying and adaptive selection on regional variation in human mtDNA. Science, 2004. 303(5655): p. 223-226.